
As cell and gene therapies (CGTs) move from experimental therapies to real-world treatments, developers face one major challenge:
How do you build a robust, reproducible manufacturing process around something as complex as living cells or gene vectors?
The answer, at least the one industry has tested, lies in Quality by Design (QbD). Before the introduction of Quality by Design (QbD), pharmaceutical development largely followed a Quality by Testing (QbT) model. In this reactive approach, product quality was verified by testing the final product, often without a deep understanding of how process variables influenced outcomes. Manufacturing changes or scale-up often introduced risk, and regulatory filings lacked the transparency to explain why processes worked—only that they did.
To modernize this model, regulatory agencies like the FDA and ICH (International Council for Harmonisation) introduced QbD in the early 2000s through ICH guidelines Q8 (Pharmaceutical Development), Q9 (Quality Risk Management), and Q10 (Pharmaceutical Quality System). The goal: move toward a proactive, science- and risk-based framework where quality is built into every step of product development and manufacturing.
QbD starts by defining the Quality Target Product Profile (QTPP)—the ideal safety, efficacy, and performance profile of the final product. Developers then identify Critical Quality Attributes (CQAs), followed by the Critical Material Attributes (CMAs) and Critical Process Parameters (CPPs) that influence them. Using experimental tools like Design of Experiments (DoE) and statistical analysis, a design space is established to define acceptable process ranges. The final output is a control strategy ensuring consistent product quality through ongoing monitoring.
Table below summarizes definitions, examples and a few references.
Term | Definition | Cell & Gene Therapy Example | Reference |
QTPP (Quality Target Product Profile) | The intended quality profile of the product—describes the ideal outcome. | • ≥80% CAR-T potency • Viability ≥70% after thaw • Transduction efficiency ≥30% • Cryo shelf life ≥12 months | |
CQA (Critical Quality Attribute) | A physical, chemical, biological, or microbiological property that must be controlled to ensure product quality. | • Vector genome copies per cell • Identity by flow cytometry • Endotoxin level <5 EU/mL • Residual plasmid DNA levels | |
CPP (Critical Process Parameter) | A variable in the process that impacts CQAs and must be controlled tightly. | • MOI (Multiplicity of Infection) • Viral incubation time • Culture conditions (O₂, CO₂, agitation) • Washing buffer composition | |
CMA (Critical Material Attribute) | A raw material property (input) that can affect CQAs and must be monitored or controlled. | • Donor T-cell viability • Vector purity and concentration • Serum or media composition• Bead-to-cell activation ratio |
This post outlines how QbD applies across four critical stages of process qualification: Engineering Runs, Pilot Runs, Comparability Runs, and Process Performance Qualification (PPQ). Along the way, we’ll explore CQAs, CPPs, CMAs, DoE, and the Control Strategy Document, with examples pulled from real FDA-reviewed BLAs like Kymriah and Zolgensma.
Engineering, Pilot, Comp, and PPQ Runs in CGT Manufacturing
In cell and gene therapy, the transition from development to commercial manufacturing relies on structured process qualification phases. Each phase generates evidence to support a robust, repeatable, and regulatory-compliant process.
Engineering runs are early development-stage batches using non-GMP or development-grade materials. Their purpose is to explore the relationship between Critical Process Parameters (CPPs) and Critical Quality Attributes (CQAs), define initial process ranges, and support assay development. These runs are essential for mapping the process design space and informing risk assessments.
In this phase, you:
- Explore design space through small-scale or non-GMP runs
- Identify relationships between CPPs and CQAs
- Test feasibility of assays, sampling, and tech transfer
Pilot runs simulate full-scale GMP conditions, using qualified materials and final equipment. They verify that the process performs consistently at scale and allow teams to finalize in-process controls, batch record formats, and material handling protocols. QbD Angle: Pilots test the “edges” of your design space — ensuring robustness.
- Use near-GMP conditions and materials
- Confirm SOPs, process timing, equipment readiness
- Finalize critical parameters for full-scale runs
Comparability runs are triggered by process changes (e.g., scale, equipment, material sources). They use side-by-side or matched-run data to confirm that CQAs remain unaffected, as required by FDA comparability guidance. Example: Zolgensma’s BLA included comparability data from multiple manufacturing facilities with matching CQA outcomes and validated assays.
PPQ runs are the final validation stage, often requiring three consecutive full-scale GMP batches. These runs demonstrate that the control strategy maintains product quality across variability in manufacturing.
- Requires ≥3 GMP lots (or justified statistical model)
- Confirms repeatability and control strategy effectiveness
- Includes IPCs, batch records, and assay results
Example: Kymriah’s BLA submitted three GMP PPQ lots that met viability, potency, and purity specs under commercial conditions.
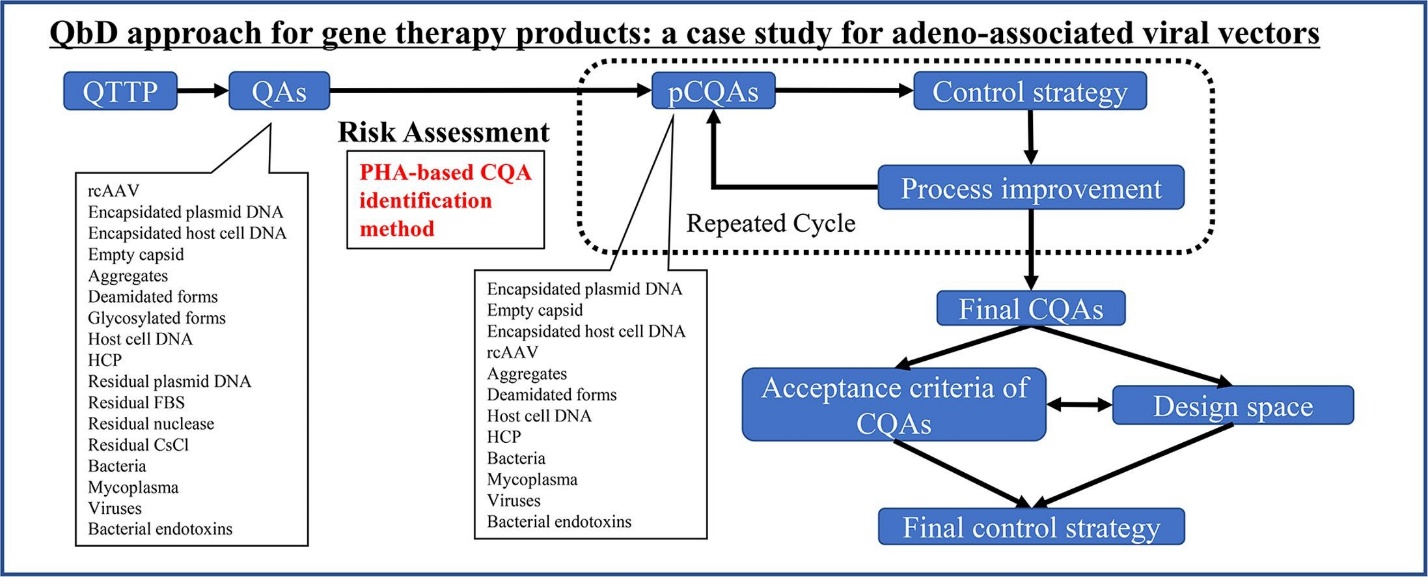
Figure 1 Optimization of the quality by design approach for gene therapy products: A case study for adeno-associated viral vectors https://doi.org/10.1016/j.ejpb.2020.08.002
Design of Experiments (DoE)
DoE is a powerful statistical tool used in the Quality by Design (QbD) framework to systematically investigate how process parameters affect product quality. Rather than changing one variable at a time (OVAT), DoE evaluates multiple variables simultaneously, allowing developers to understand interactions, optimize performance, and build robust manufacturing processes.
In the context of QbD, DoE supports the identification and control of Critical Process Parameters (CPPs) that impact Critical Quality Attributes (CQAs). For example, in cell therapy, DoE might be used to examine how MOI, incubation time, and media composition together influence transduction efficiency and cell viability. Common DoE methods include factorial designs, response surface methodology (RSM), and Plackett-Burman screening designs. These help define the design space—a multidimensional region of operating conditions shown to yield acceptable product quality. DoE not only accelerates development by reducing the number of experiments needed, but it also provides statistically meaningful insights to justify control strategies during regulatory submissions. When implemented early, DoE de-risks scale-up, tech transfer, and PPQ by ensuring that the process is data-driven and predictable, aligning with ICH Q8 and FDA expectations.
The Control Strategy Document
The Control Strategy Document serves as the regulatory and operational backbone in cell and gene therapy manufacturing, aligning your process with quality expectations from agencies like the FDA. Take CAR T as an example, According to the FDA’s “Considerations for the Development of CAR T Cell Products”, sponsors must clearly define Critical Quality Attributes (CQAs) and design Critical Process Parameters (CPPs) to consistently safeguard those CQAs. The document should include:
- In‑process controls (IPCs) and release criteria, such as viability thresholds and transgene expression assays.
- Analytical comparability plans for process or material changes, a key element emphasized in the guidance
- Continuous chain-of-identity and stability assurances, including consistency in leukapheresis procedures, vector titer characterization, and process control strategies
Together, these FDA guidelines inform a Control Strategy Document that integrates CQAs, CPPs, IPCs, assay validation, stability plans, and comparability frameworks—organized to ensure safe, reproducible, and compliant CGT manufacturing.
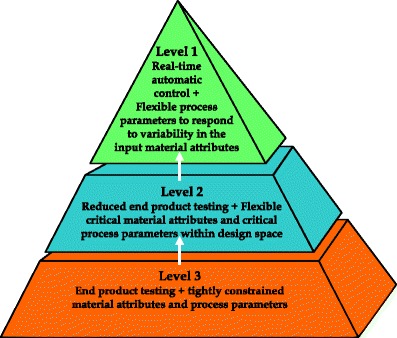
Figure 2 Control strategy implementation options from doi: 10.1208/s12248-014-9598-3
Final Thoughts
QbD isn’t just regulatory jargon. For cell and gene therapy, it’s your blueprint for making something reproducible from something inherently variable. From first engineering runs to PPQ filing, QbD offers a powerful structure for building—and defending—your process.
If there is a topic that you would like to see here or have a question, please drop us a line at hello@assay.dev